
Protocol for the simulation of electron binding energies

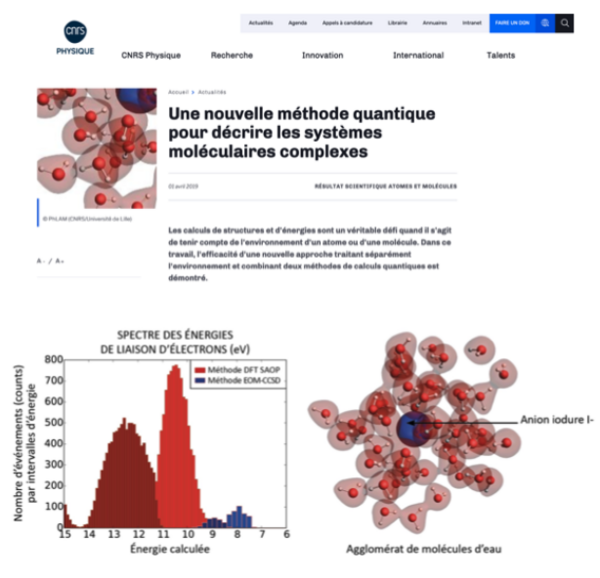
As part of the PhD work of Y. Bouchafra, we combined developments carried out within the group (relativistic correlated electronic structure, quantum embedding, and classical polarizable force fields for charged species) to simulate the binding energies of valence electrons of halides in small water droplets.
Electron binding energies are a key quantity to characterize processes taking place in bulk and at interfaces. Taking into account relativistic effects in our RCC-in-DFT calculations enabled the simulation of halide ions from fluoride to iodide, and allowed us to achieve very high accuracy at a modest computational cost compared to more sophisticated (and computationally expensive) competing approaches.
Our protocol also allowed us to investigate astatide (At-), an element with potential for use in radiotherapy but whose relatively short lifetime makes experimental studies challenging.
This work was selected as Editor’s suggestion by Physical Review Letters (DOI: 10.1103/PhysRevLett.121.266001), and has been highlighted by CNRS Physics in 2019.
This study also paved the way, after additional developments (the implementation of the core-valence separation approximation in our relativistic EOM-CCSD codes), for simulating core electron binding energies, such as those measured by X-ray photoelectron spectroscopy (XPS). We have carried out simulations of XPS spectra for chloride and HCl on droplets and on ice surfaces (DOI: 10.1039/D1CP05836C) as part of the PhD of R. Opoku, and of the uranyl tetrachloride species (UO2Cl42-) in a host Cs2UO2Cl4 crystal (arXiv:2302.07223) as part of the PhD of W. Misael.