The last publication of PCMT honored by Physical Review Letters.
PCMT Vie du labo Parutions
The publication "Predictive Simulations of Ionization Energies of Solvated Halide Ions with Relativistic Embedded Equation of Motion Coupled Cluster Theory." of PCMT Team (Yassine Bouchafra, Avijit Shee, Florent Réal, Valérie Vallet, and André Severo Pereira Gomes) has been selected by the publisher Physical Review Letters.
Abstract of the article Phys. Rev. Lett. 121, 266001 published on December 28, 2018 : Link
Predicting binding energies of complex systems via efficient molecular simulations.
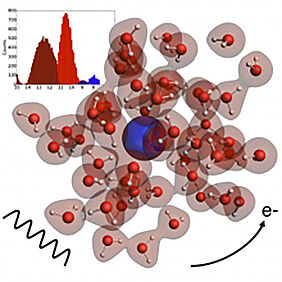
Physico-chemical processes at the nanoscopic scale usually involve the changes in internal states the electrons in atoms, molecules and materials. In order to better understand such processes, detailed information on the binding energies of the electrons in each of these systems, when they are separated or when they interact with each other are needed. A way to do so is via experimental techniques such as photoelectron spectroscopy, which determines the energy levels of the electrons through their ejection from a particular system. As such experiments can be difficult to interpret, theory and computational modeling can come to the rescue and provide ways to construct models to the actual physics system. Theorists should provide models which are as accurate (i.e. reproduce faithfully the experimental results), predictive (i.e. can tell experimentalists what should be happening in a system they haven’t yet measured), and computationally efficient (i.e. to be able to provide answers relatively quickly) as possible, while being as easy to understand from a physical point of view as possible. In our work we introduce a promising computational approach for calculating binding energies which meets the criteria above, and have applied it to study the effect of solvation on the binding energies of halogen anions, from fluoride to iodide, relevant to atmospherical issues, and then on to astatide – a radioactive element which shows promise in nuclear medicine applications, but which is very difficult to study experimentally due to its short half-life. Our simulations consider the dynamics of each of the halides in small water droplets (see figure), and are unique in that they easily incorporate so-called relativistic effects – arising due to the increase the atomic mass of the nuclei as one moves down in the periodic table towards astatide, which makes the electrons near the nuclei move at speeds very close to the speed of light and with it change how the whole system behaves – as well as can capture the binding energies both of the halides and the water molecules surrounding the halides at the same time. © 2018 Y. Bouchafra, A. Shee, F. Réal, V. Vallet, and A. Gomes All rights reserved.
Physico-chemical processes at the nanoscopic scale usually involve the changes in internal states the electrons in atoms, molecules and materials. In order to better understand such processes, detailed information on the binding energies of the electrons in each of these systems, when they are separated or when they interact with each other are needed. A way to do so is via experimental techniques such as photoelectron spectroscopy, which determines the energy levels of the electrons through their ejection from a particular system. As such experiments can be difficult to interpret, theory and computational modeling can come to the rescue and provide ways to construct models to the actual physics system. Theorists should provide models which are as accurate (i.e. reproduce faithfully the experimental results), predictive (i.e. can tell experimentalists what should be happening in a system they haven’t yet measured), and computationally efficient (i.e. to be able to provide answers relatively quickly) as possible, while being as easy to understand from a physical point of view as possible. In our work we introduce a promising computational approach for calculating binding energies which meets the criteria above, and have applied it to study the effect of solvation on the binding energies of halogen anions, from fluoride to iodide, relevant to atmospherical issues, and then on to astatide – a radioactive element which shows promise in nuclear medicine applications, but which is very difficult to study experimentally due to its short half-life. Our simulations consider the dynamics of each of the halides in small water droplets (see figure), and are unique in that they easily incorporate so-called relativistic effects – arising due to the increase the atomic mass of the nuclei as one moves down in the periodic table towards astatide, which makes the electrons near the nuclei move at speeds very close to the speed of light and with it change how the whole system behaves – as well as can capture the binding energies both of the halides and the water molecules surrounding the halides at the same time. © 2018 Y. Bouchafra, A. Shee, F. Réal, V. Vallet, and A. Gomes All rights reserved.